
What makes psoriasis sore: Novel role of immune system in the disease
More than 130 million people around the globe suffer from psoriasis vulgaris, a chronic condition characterized by skin inflammation, scales, and dry patches. However, its pathology is not fully clear. In a recent study, dermatology researchers at Fujita Health University have uncovered a complex cellular mechanism responsible for the onset of psoriasis and highlighted potential therapeutic targets for future treatment.
Psoriasis has always been a common disease. Historically, its causes were obscure and surrounded by stigma; it wasn’t until recently that scientists categorized it as an autoimmune condition. Indeed, modern scientific research shows that the body’s own T-cells, macrophages and dendritic cells are responsible for attacking healthy skin tissue, triggering inflammation and proliferation of skin cells, and resulting in the characteristic red, painful plaque-like lesions experienced by psoriasis patients.
But although these immune-mediating cells have been identified as the primary culprits for the breakdown of healthy skin, their roles do not fully clarify the underlying cause. What makes these cells behave so abnormally?
Based on their existing knowledge of the cellular and genetic pathways linked with the disease, Professor Kazumitsu Sugiura and Dr. Soichiro Watanabe from Fujita Health University, Japan, along with their colleagues, attempted to find out. Their study helped them clarify the role of another potential culprit in the formation of psoriasis lesions: neutrophils. Their findings are published in Scientific Reports.
The most common type of white blood cell, neutrophils are known to be associated with various other chronic and autoimmune conditions, such as rheumatoid arthritis and lupus. Neutrophils have pro-inflammatory effects: once activated, they travel to the site of infection and effectively undergo cell death. During this process, they burst open and release their contents, forming what scientists call “neutrophil extracellular traps” (NETs), a web of proteins and genetic information from the nucleus of the former cell.
NETs have reportedly been found in psoriasis lesions. However, the mechanisms linking the formation of NETs and severe skin inflammation are unknown. Prof Sugiura explains: “NETs and neutrophils can induce inflammation through various mechanisms; we aim to clarify the role of NET signaling and possibly prevent the development of severe psoriasis-like lesions”.
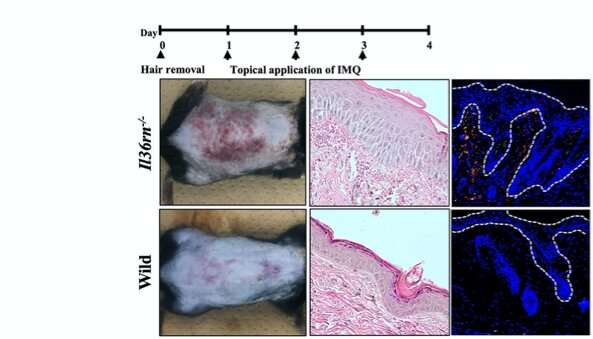
A precursor to the activation of NETs in psoriasis is a group of signaling proteins called IL-36 cytokines, whose role in recruiting various white blood cells and inducing inflammation is well acknowledged. For instance, a mutation in the Il36rn gene that causes a deficiency of the IL-36 receptor antagonist (IL-36Ra) protein, allows IL-36 to act uninhibited, leading to inflammation, inducing the proliferation of epidermal cells and increasing neutrophil count in psoriasis-like lesions in the mutant mice, in a condition known as DITRA. This results in skin cells keratinizing (hardening) in the manner seen in psoriasis lesions.
Armed with this theoretical knowledge, Prof. Sugiura and team studied two groups of mice: one healthy (wild type) and another with the aforementioned Il36rn gene mutation. They induced psoriasis-like lesions using imiquimod, a drug known to stimulate IL-36 signaling and subsequent immunological response. After examining the lesions, they found that mutant mice suffered more severe inflammation than the wild type. A more detailed histological analysis revealed that the mutant mice also had significantly higher neutrophil counts and NET concentrations than the wild type. The scientists went one step further and examined the mRNA levels of target cytokines known to be involved in psoriasis formation to elucidate the IL-36 pathway, thereby linking cause and effect.
Having identified these complex cellular mechanics, the scientists made a list of potential targets to block the inflammation pathway and prevent psoriasis. They injected the diseased mice with Cl-amidine, a drug which specifically targets and inhibits PAD4—an enzyme responsible for the formation of NETs. After only three days of treatment, they found the lesions, as well as the levels of inflammatory cells and IL-36 signaling proteins, significantly reduced.
Source: Read Full Article