
Study suggests algorithm may help optimize future COVID-19 vaccine design
Scientists predict the COVID-19 pandemic is on its way to becoming endemic. The rising spread of COVID-19 variants such as Delta has helped infect unvaccinated and some vaccinated individuals. COVID-19 vaccines continue to be the best way to protect against severe illness and death, but there may be a need for vaccines to protect against a specific variant.
Research led by Vivek R. Nerurkar from the University of Hawaiʻi at Mānoa developed a mathematical algorithm that can help optimize the vaccine platform. The algorithm monitors the evolution of SARS-CoV-2 lineages and incorporates information such as spike mutations from variants of concern, SARS-CoV-2 genomes, and clinical samples.
The researchers write:
Our findings have relevance to the future of tracking SARS-CoV-2 and of SARS-CoV-2 vaccine design…If established in real-time, the herein described algorithm will allow researchers to understand the evolving SARS-CoV-2 genome preemptively rather than responsively.”
The study “Algorithm for the Quantitation of Variants of Concern for Rationally Designed Vaccines Based on the Isolation of SARS-CoV-2 Hawaiʻi Lineage B.1.243” is published on the bioRxiv* preprint server, while awaiting peer review.
.jpg)
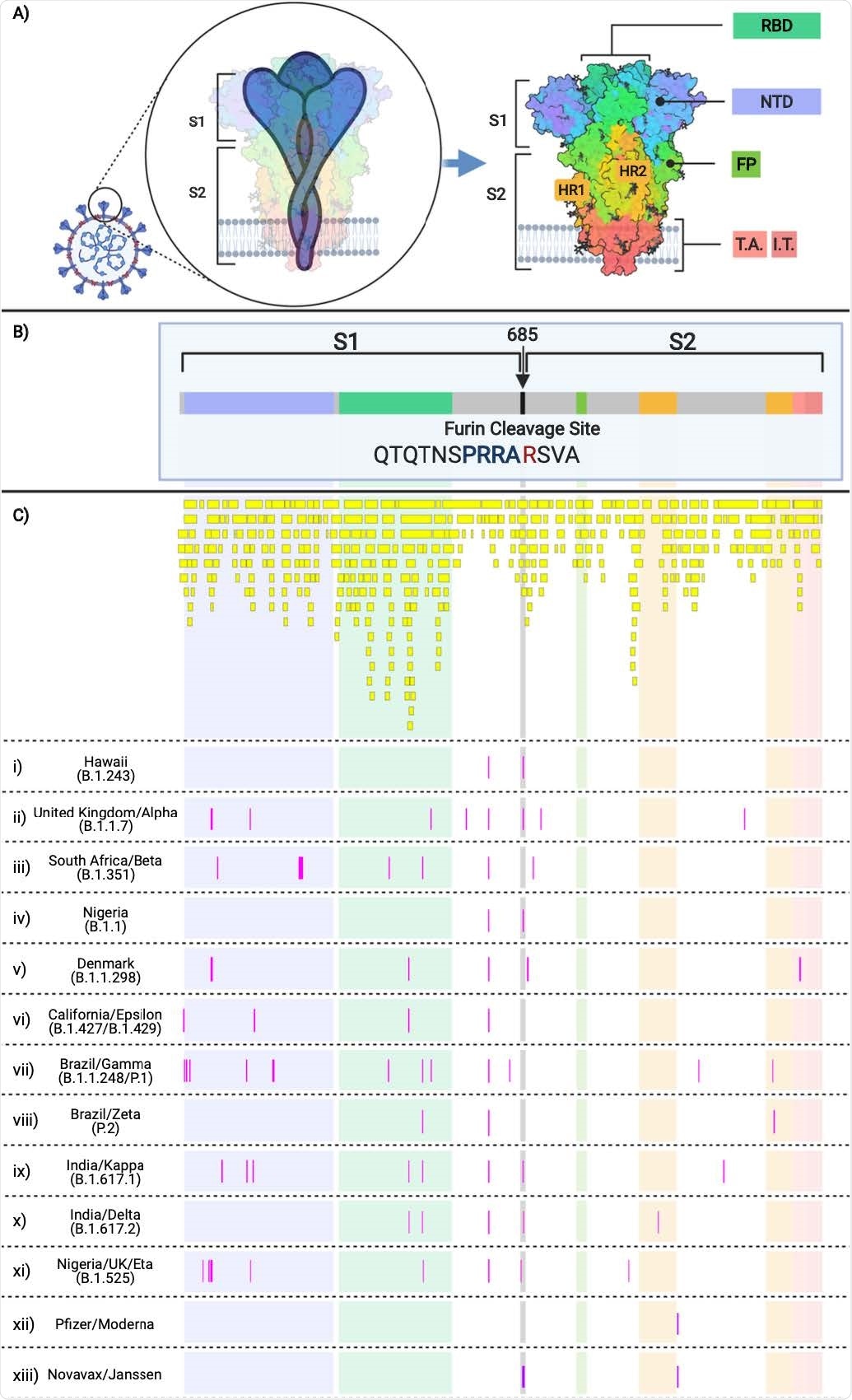
Validating the algorithm using SARS-CoV-2 isolates from the B.1.243 lineage
The researchers applied an adaptive algorithm to monitor SARS-CoV-2 evolution and other genetic changes. The goal was to use the information to help design logical next-generation vaccines.
They collected SARS-CoV-2 samples 5 days after two patients tested positive for SARS-CoV-2. Genomic sequencing was performed and compared to data from both GenBank and GISAID to identify the SARS-CoV-2 lineage. The samples were found to be related to the B.1.243 lineage.
The B.1.243 variant once made up 72% of genomic sequences from Hawaii. However, by July 28, 2021, the variant decreased to about 23% of all sequences. At the time of the study, the Delta variant was taking over as the dominant SARS-CoV-2 strain in the area.
The SARS-CoV-2 genome evaluation showed 49 mutations in the SARS-CoV-2 isolates related to the B.1.243 lineage. About 9 of the spike protein mutations observed had a 70% prevalence worldwide. The 10th spike protein mutation was D614G, which is widespread in more than 99% of strains.
Isolating SARS-CoV-2 from samples and analyzing published sequences showed that B.1.243 made up more than 40% of all cases in Hawaii. However, the researchers note that it is unlikely to be considered a variant of concern given its decreasing prevalence worldwide.
B.1.243 helped validate the algorithm and analyze variants of concern/interest based on emerging spike protein amino acid changes for future surveillance and vaccine design.
The algorithm provided numerical value to each variant of concern and predicted the likelihood it would spread across the population. It also calculated the probability each variant of concern would undertake future mutations.
Spread of variant of concerns, especially Gamma, are projected to increase worldwide
The research team used the data to predict exponentially emerging mutation to the SARS-CoV-2 isolates in the current study.
The Centers for Disease Control and Prevention currently identifies Alpha, Beta, Delta, and Gamma as four variants of concern.
Results suggest all four variants of concern will have an exponential spread, with Gamma being the most dominant worldwide.
“The Delta variant, predicted to be exponentially emerging by this quantitative analysis with an r-value of 0.96 as of April 2021 at 1% prevalence, has become the most prevalent worldwide as of June 2021, representing 64% of worldwide sequences,” wrote the team.
Based on the findings, the team suggests the algorithm can serve to monitor for SARS-CoV-2 mutations and serve as a baseline for choosing the primary structure of vaccines. They also argue that it’s a valuable tool in taking a proactive and predictive approach to dealing with future variants.
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- Maison DP, et al. Algorithm for the Quantitation of Variants of Concern for Rationally Designed Vaccines Based on the Isolation of SARS-CoV-2 Hawaiʻi Lineage B.1.243. medRxiv, 2021. doi: https://doi.org/10.1101/2021.08.18.455536, https://www.biorxiv.org/content/10.1101/2021.08.18.455536v1
Posted in: Device / Technology News | Medical Research News | Disease/Infection News
Tags: Amino Acid, Coronavirus Disease COVID-19, Evolution, Genetic, Genome, Genomic, Genomic Sequencing, Mutation, Pandemic, Protein, Quantitative Analysis, Research, SARS, SARS-CoV-2, Spike Protein, Vaccine

Written by
Jocelyn Solis-Moreira
Jocelyn Solis-Moreira graduated with a Bachelor's in Integrative Neuroscience, where she then pursued graduate research looking at the long-term effects of adolescent binge drinking on the brain's neurochemistry in adulthood.
Source: Read Full Article