
Researchers develop random priming amplification method for whole-genome sequencing of SARS-CoV-2 and H1N1
Recent advances in next‐generation sequencing (NGS) such as targeted or non-targeted whole-genome sequencing (WGS) and computational analyses that can efficiently process large amounts of data have enabled comprehensive analyses of viral genomes in clinical and research settings. For example, these techniques have been applied to identify causative agents and origins of outbreaks, monitor transmissions, or study the dynamics of epidemics such as Ebola, Zika, and the ongoing severe acute respiratory syndrome coronavirus 2 (SARS‑CoV‑2) pandemic.
“Whole-genome sequencing (WGS) is increasingly applied in clinical medicine as it has the potential to identify clinically actionable genetic variants informing early medical intervention.”
The first SARS‐CoV‐2 genomes recovered in early 2020 from 8 patients in Wuhan showed an identity of 99.98% and this high level of shared genomic similarity in viral genomes showed that it is a new virus that has not been circulating for a long time in humans and was possibly a spillover from animals to humans.
Recent studies showed that intra-patient SARS-CoV-2 genetic diversity increased after plasma treatment of patients having high viral loads. Moreover, a high rate of SARS-CoV-2 genome mutation was detected in immunodeficient patients infected with the virus.
Non-targeted whole-genome sequencing is a potent tool that helps comprehensive identifications of constituents of microbes in a sample. Since there is no need to direct the analysis to any kind of identification before genome sequencing, this technique can decrease bias and false-negative results. It also allows the determination of genetic aberrations such as single nucleotide variants, insertions, deletions, and copy number variants, even in the noncoding protein regions of the genome.
Comparing the performance of 4 different random priming amplification methods to recover SARS-CoV-2 RNA
Researchers from the UK recently compared the performance of 4 different random priming amplification methods to recover SARS-CoV-2 RNA. In method 1 (H-P), the reverse transcriptase (RT) step included random hexamers, whereas, in methods 2-4, RT incorporated an octamer primer with a known tag. This study is published on the bioRxiv* preprint server.
In methods 1 and 2, (K-P) sequencing was performed on material derived from the RT-PCR step, while in methods 3 (SISPA) and 4 (S-P), additional amplification was performed before sequencing. They also presented the S-P technique that might be applicable for Oxford Nanopore real-time sequencing as a non-targeted primer-based protocol is not available yet.
“Presented in this study, the SISPA protocol allowed for whole-genome assembly of both viruses using only one primer in a sequence-independent reaction.”
SISPA method is the most efficient method of the 4 methods for whole-genome sequencing of SARS-CoV-2
The results showed that the SISPA method described by the authors in this study was the most efficient method for non-targeted/random priming whole-genome sequencing of SARS-CoV`-2. This method allowed for whole-genome assembly of SARS-CoV-2 and influenza A(H1N1) pdm09 in mixed samples. They determined the detection limit – 103 pfu/ml (Ct, 22.4) for whole-genome assembly – and the limit of characterization of SARS-CoV-2 – 101 pfu/ml (Ct, 30) for metagenomics detection. The whole-genome sequences obtained using the SISPA (or S-P) method in this study were free of primer bias and allowed for polymorphism analysis.
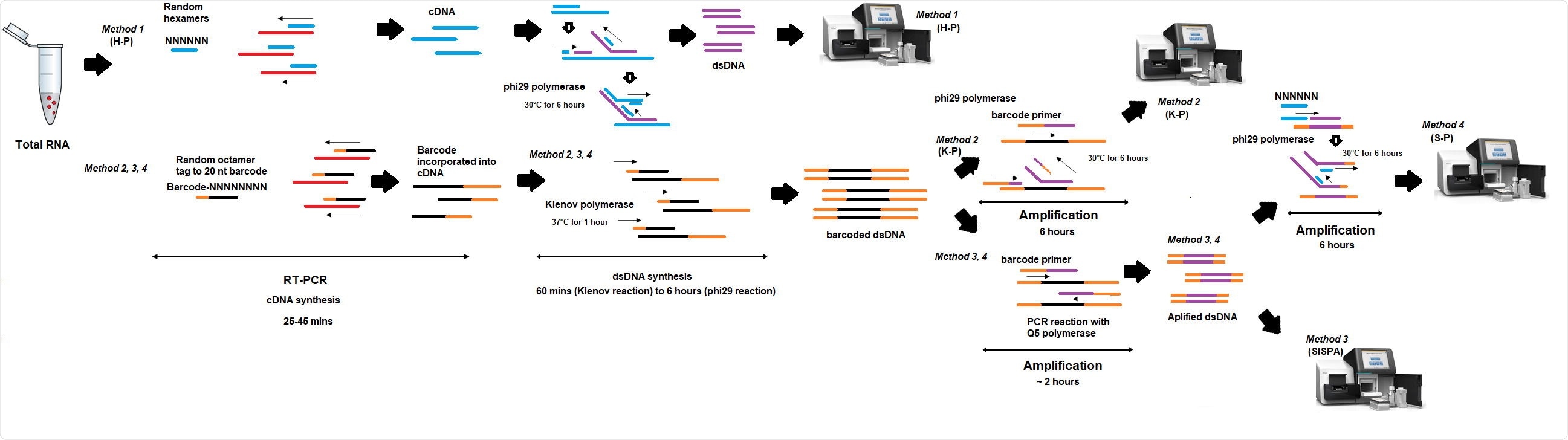
The SISPA method is mainly used to obtain genome sequences of RNA viruses or investigating complex clinical samples with mixed infections in a single reaction since no sequence information is required.
According to the authors, this method can be used to monitor genomic changes and evolution of the virus, for fast and simple metagenomics detection, and to assess the pathogen profile of the sample that might help identify other factors that might be involved in the coronavirus disease (COVID-19) infection.
“Previous studies have shown active genetic recombination events in SARS-CoV-2 genomes which may reduce the accuracy of conventional qRT-PCR detection, and thus the primers should be precisely chosen to address these challenges.”
*Important Notice
bioRxiv publishes preliminary scientific reports that are not peer-reviewed and, therefore, should not be regarded as conclusive, guide clinical practice/health-related behavior, or treated as established information.
- A random priming amplification method for whole genome sequencing of SARS-CoV-2 and H1N1 influenza A virus Klaudia Chrzastek, Chandana Tennakoon, Dagmara Bialy, Graham Freimanis, John Flannery, Holly Shelton bioRxiv 2021.06.25.449750; doi: https://doi.org/10.1101/2021.06.25.449750, https://www.biorxiv.org/content/10.1101/2021.06.25.449750v1
Posted in: Device / Technology News | Medical Research News | Disease/Infection News
Tags: Coronavirus, CT, Evolution, Genetic, Genetic Recombination, Genome, Genomic, H1N1, Illumina, Influenza, Library Preparation, Medicine, Metagenomics, Mutation, Nucleotide, Pandemic, Pathogen, Polymerase, Protein, Research, Respiratory, Reverse Transcriptase, RNA, SARS, SARS-CoV-2, Severe Acute Respiratory, Severe Acute Respiratory Syndrome, Syndrome, Virus

Written by
Susha Cheriyedath
Susha has a Bachelor of Science (B.Sc.) degree in Chemistry and Master of Science (M.Sc) degree in Biochemistry from the University of Calicut, India. She always had a keen interest in medical and health science. As part of her masters degree, she specialized in Biochemistry, with an emphasis on Microbiology, Physiology, Biotechnology, and Nutrition. In her spare time, she loves to cook up a storm in the kitchen with her super-messy baking experiments.
Source: Read Full Article